Dissecting the requirements for specific antioxidant enzymes for ROS resistance
NRF2 is a stress-responsive transcription factor that directs select transcriptional programs in response to oxidative stimuli. NRF2 levels are tightly controlled by KEAP1, which directs NRF2 destruction. Our early work demonstrated a critical role for K-Ras induced NRF2 transcription in lung and pancreatic tumor initiation (DeNicola et al. Nature 2011). Further, NRF2 and KEAP1 mutations are common in certain cancers, including non-small cell lung cancer, and lead to loss of NRF2 degradation and constitutive NRF2 accumulation, thereby promoting glutathione synthesis, detoxification of reactive oxygen species (ROS) and proliferation. Because NRF2 engages both the glutathione- and thioredoxin-dependent antioxidant systems, it raises the question of whether any these semi-redundant systems can be targeted to reverse the resistance to ROS conferred by NRF2 activation. We find that the thioredoxin system or superoxide dismutase 1 are required for the resistance to the ROS-generating molecule ß-lapachone, but the glutathione synthesis and catalase are dispensible (Torrente et al., Redox Biology 2020). Moreover, using a CRISPR screening approach, we found that targeting enzymes within the thioredox system and the pentose phosphate pathway could sensitize KEAP1 mutant cells to this molecule (Jiang et al., Redox Biology 2022). Interestingly, we also found that deleting the mitochondrial superoxide dismutase 2 could also sensitize to ß-lapachone due to a failure to maintain ATP and NADPH.
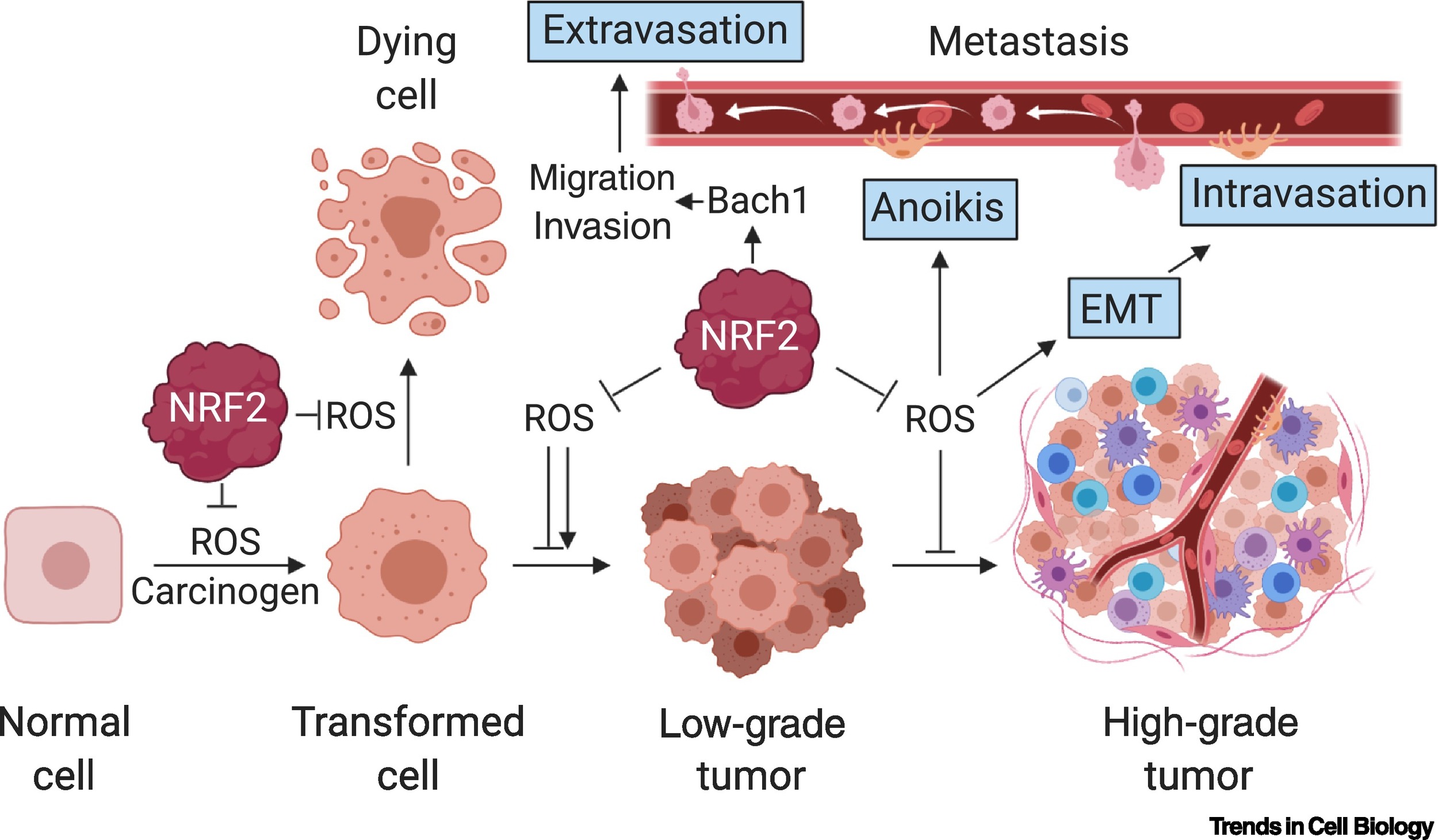
Understanding the complex roles of NRF2 in tumor progression and metastasis
However, we find that the effects of NRF2 on tumor phenotypes are complex and highly context dependent. In a collaborative study with the Vousden lab, we showed that active suppression of ROS by NRF2 suppresses pancreatic cancer metastasis (Cheung et al., Cancer Cell 2020). To assess the effects of NRF2 activation on cellular metabolism and tumorigenesis in an isogenic system, we generated genetically engineered mouse models of both the NRF2D29H and KEAP1R554Q mutations found in human NSCLC. Surprisingly, these mouse models demonstrated that NRF2 activation promotes tumor initiation but impairs the progression of KrasG12D lung tumors (Kang et al. eLife 2019, DeBlasi et al. Cancer Research 2023). Work is ongoing to understand how NRF2 activation impairs tumor progression.
While NRF2 activation promotes lung tumor initiation and early progression in murine models of lung cancer, the specific NRF2 targets that mediate these phenotypes remain poorly understood. NRF2 regulates two parallel antioxidant systems mediated by thioredoxin reductase 1 (TXNRD1) and glutathione reductase (GSR), which promote the reduction of protein antioxidant thioredoxin (TXN) and tripeptide antioxidant glutathione (GSH), respectively. To dissect the roles of these systems, we deleted TXNRD1 and GSR alone, or in combination, in lung tumors harboring mutations in KrasG12D and Nrf2D29H. Our findings revealed that these antioxidant systems play distinct, stage-specific roles in tumorigenesis (Sherwood et al. Redox Biol 2025). Tumor initiation was promoted by GSR expression, but not TXNRD1, regardless of Nrf2 status. In contrast, Nrf2D29H tumors, but not Nrf2WT tumors, were dependent on TXNRD1 for tumor progression, while GSR was dispensable at this stage. Simultaneous deletion of both GSR and TXNRD1 reduced both initiation and progression independent of Nrf2 status, but surprisingly did not completely abrogate tumor formation, suggesting additional compensatory mechanisms. These findings demonstrate that the thioredoxin and glutathione antioxidant systems have unique, non-redundant roles in different stages of tumor development, providing new insights into how NRF2 promotes cancer progression and identifying potential stage-specific therapeutic vulnerabilities.
Open Questions: