Overview
Tumors can be viewed as complex eco-systems, where evolving populations of genetically and phenotypically heterogeneous malignant cells are engaged in a network of reciprocal interactions with multiple normal cell types and non-cellular components of tumor micro and macro environment. Mainstream reductionistic molecular oncology studies have revealed molecular mechanisms responsible for malignant phenotypes (aka “hallmarks of cancers”) in great mechanistic detail, enabling the development of highly effective targeting therapies. Unfortunately, these therapies are rarely curative, as, owing to microenvironmental complexity and tumor heterogeneity they fail to eliminate all of the tumor cells, allowing cancers to evolve adaptations to therapy induced selective pressures, ultimately resulting in relapsed disease. Thus, despite the spectacular developments in understanding proximal mechanisms of the disease, this knowledge has not yet resulted in dramatic improvements of clinical outcomes. We believe that in order to break the impasse, we need to account for tumor complexity, which can be achieved by combining knowledge of proximal mechanisms with an eco-evolutionary framework to understand how tumors change in space and time.
Deciphering evolutionary trajectories leading to resistance toward ALK targeting therapies in lung cancers.
Targeted therapies directed against abnormal kinases represent the pinnacle of molecular oncology research. When matched properly with susceptible tumors, they produce strong clinical responses with much lower toxicity compared to “carpet bombing” chemotherapeutic agents. Unfortunately, in the vast majority of cases, targeted therapies eventually fail, as tumors evolve under therapy-imposed selective pressures, culminating in outgrowth of therapy-resistant tumor sub-populations. In contrast to massive research efforts directed into characterization of the end-products of evolution of acquired resistance, relatively little is known about how resistance develops. This gap of knowledge is at least partially attributable to a common assumption that evolution of resistance can be reduced to outgrowth of clones that have acquired resistance through a mutation occuring either before or during therapy. However, our studies in models of EML4-ALK lung cancers suggest a more complicated scenario. We found that the resistance originates from cells that, despite reduced sensitivity relative to the majority of cells, can barely tolerate the treatments and are much more sensitive to the drugs compared to the end-products of evolution of resistance. Rather than resulting from a binary (epi) mutational switch, development of resistance involves acquisition of multiple resistance – associated molecular changes that appear to have additive or synergistic effect, culminating in phenotypes with complete or near-complete therapy resistance. Notably, different ALK TKIs induce different evolutionary trajectories, ending with resistant cells that, despite cross-resistance to different ALK TKIs display distinct phenotypic changes and distinct patterns of cross-sensitivities.

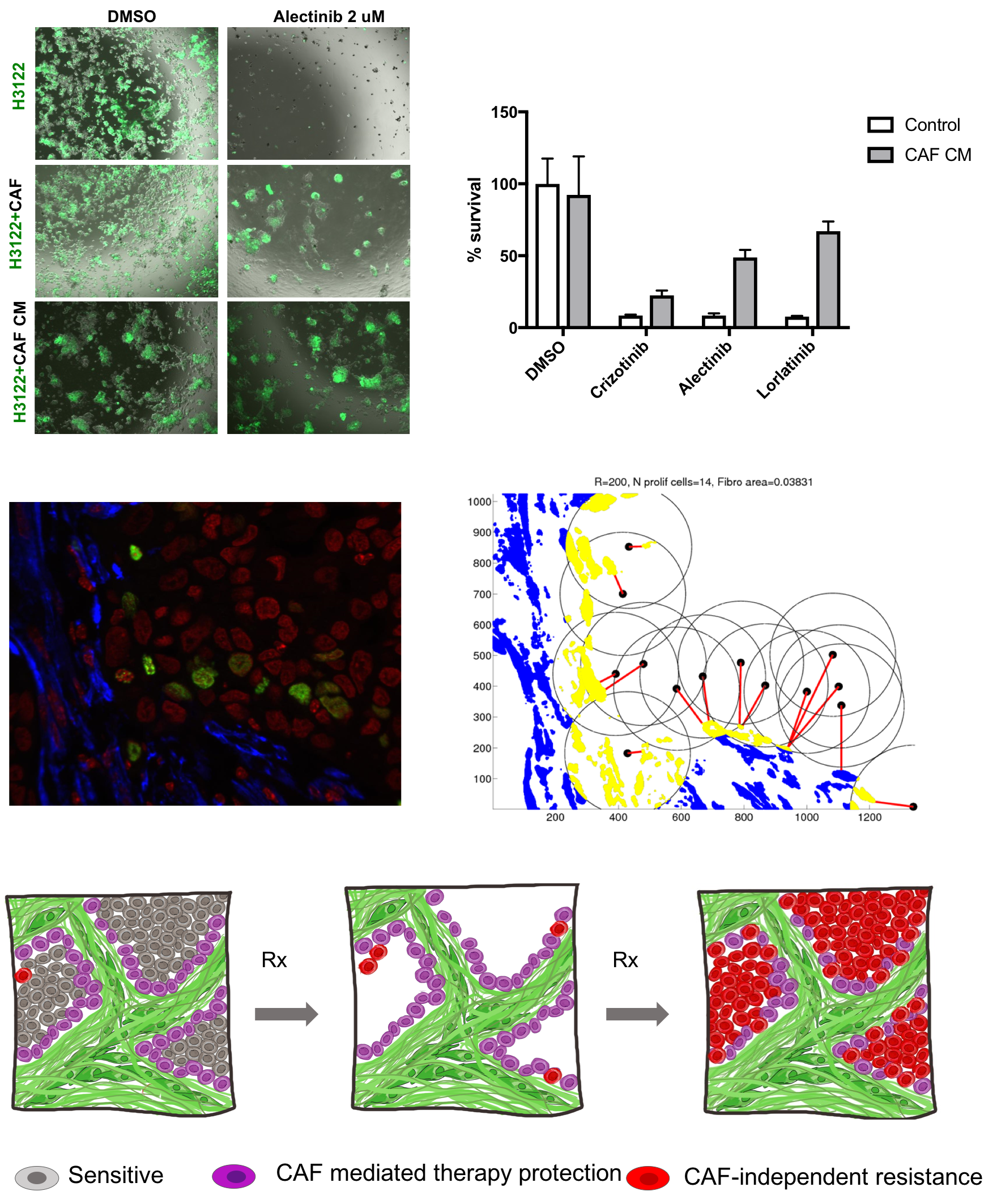
Microenvironmentally mediated therapy resistance.
Most anti-cancer therapies fail to eliminate all of the tumor cells, allowing populations of cells comprising residual disease to evolve therapy resistance. In part, the ability of therapy naive tumor cells to survive complete elimination reflects intra-tumor heterogeneity and presence of sub-populations less sensitive to the treatment. However, microenvironmental protective niches emerge as another important source of residual disease. Our work, as well as work from many other groups, has found that cancer associated fibroblasts, a major cellular component of tumor microenvironment, as well as the primary producer of extracellular matrix, can provide a striking protection against some cytotoxic and targeted therapies. Despite the mounting evidence for the importance of microenvironmental therapy resistance, potential impact of stromal protection is typically ignored during clinical diagnostics and therapeutic decision-making. We are developing methods to assess the contribution of fibroblast-mediated protection for therapy resistance in vivo, and investigating strategies to overcome this protection in breast and lung cancers.
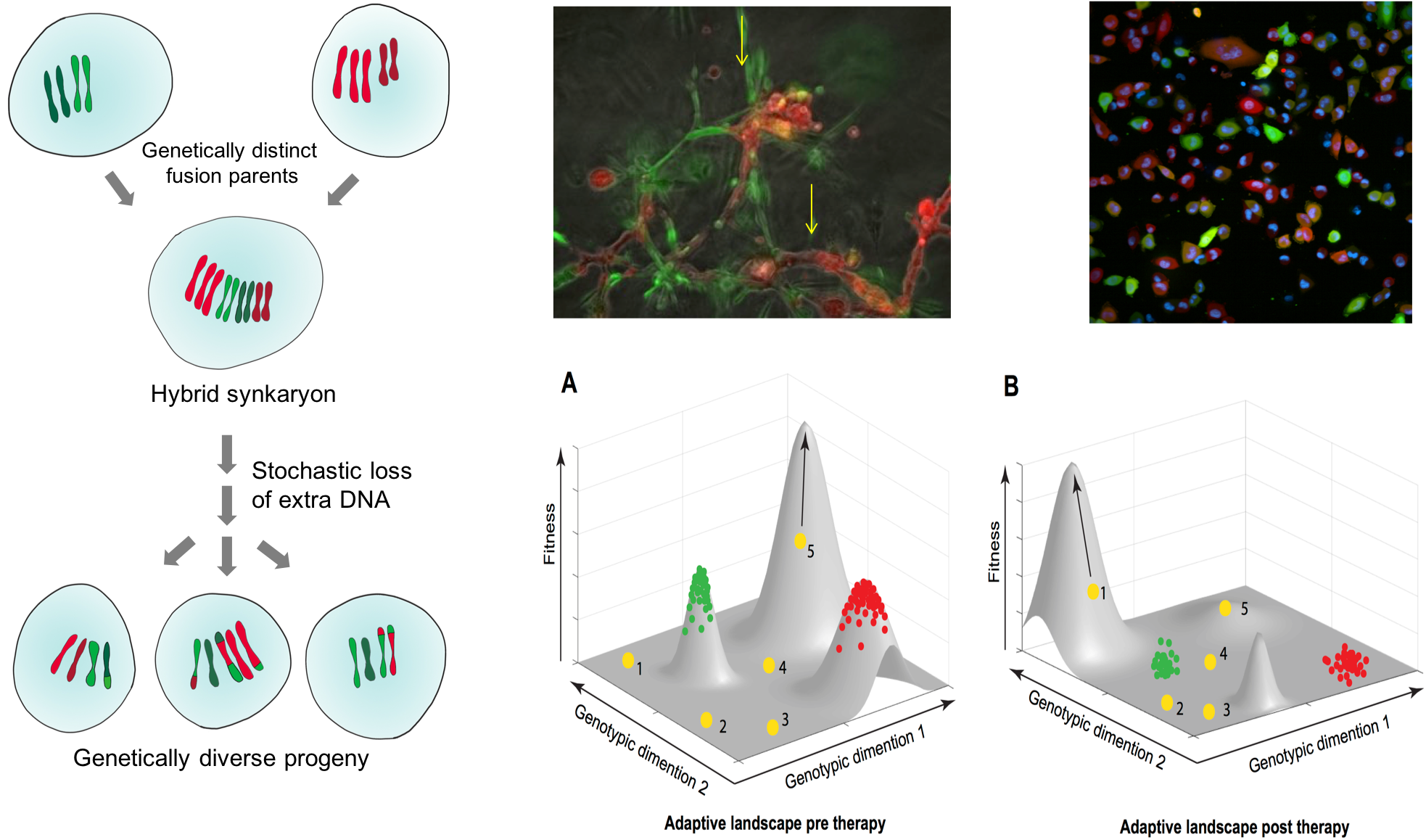
Cell fusions as a novel source of intra-tumor heterogeneity.
Ability of tumor cells to evolve is essential for tumor progression and therapy resistance, and this evovlability hinges on generating heritable diversity within populations of tumor cells. In virtually all strata of life, sexual recombination or its para-sexual analogues represent a powerful, evolutionary essential diversification mechanism. In contrast, no such processes involving recombination of DNA from distinct cells have been described for tumor cells. We and others have noticed that tumor cells can spontaneously fuse between themselves, as well with some cells from tumor microenvironment. Further, we find that some of the fusion products can generate viable clonogenic progeny. Karyotypes of hybrid cells are relatively unstable, and most of the progeny returns to the lower ploidy status. This ploidy reduction is accompanied with random or semi-random loss of genetic material from parental genomes. Thus, cell fusions followed by ploidy reduction can constitute a novel, unappreciated source of the ability of tumor cells to evolve. Despite low frequency of the fusion events, mathematical modeling performed in the lab of our collaborator David Basanta suggests that cell fusions can have oversized impact on diversity of tumor cell populations. We are currently investigating the functional importance of this diversification and are searching for approaches to limit it.