
Pancreatic cancer is a progressive, highly metastatic disease with the worst overall survival rate of any cancer in the US. The genomic landscape of pancreatic ductal adenocarcinomas (PDAC) is marked by few recurrent mutations and chromosomal instability. Somatic oncogenic mutations in KRAS are initiating driving events, followed by loss of tumor suppressor genes CDKN2A, TP53 and SMAD4. Defining additional cooperating genetic events that contribute to cancer progression and metastasis has proven to be challenging, due in part to the inter- and intra-tumor heterogeneity of genomic aberrations in primary tumors, marked by hundreds of infrequent mutations and a high propensity of gene copy number alterations (CNAs).
In an effort to identify genes that cooperate with oncogenic KrasG12D to drive PDAC in a mouse model, we performed a forward genetic screen using Sleeping Beauty (SB) insertional mutagenesis (Mann et al., PNAS 2012). The SB_PDAC model recapitulates all the histological hallmarks of human PDAC, including metastasis to the liver, peritoneum and lung, and is defined by hundreds of genes statistically enriched for SB insertions, indicative of positive selection. While there is statistical enrichment for SB_PDAC candidate driver genes with human homologs contain missense mutations in PDAC, the majority of SB_PDAC candidate drivers are not mutated in human PDAC genomes. Rather, these genes are differentially expressed, subject to CNAs and/or participate in biological pathways and processes associated with PDAC, including TGF-beta signaling, chromatin remodeling, axon guidance and RNA processing.
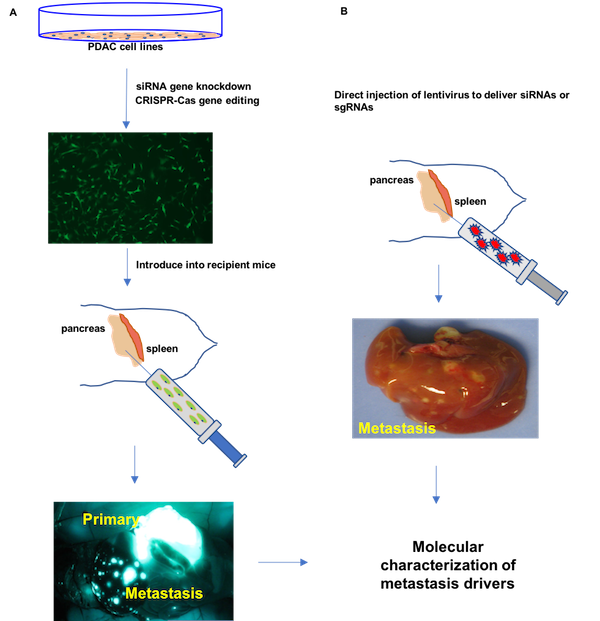
We have validated a number of novel pancreatic cancer drivers that contribute to primary tumor growth and metastases using orthotopic mouse models. Comparative genomic analysis of our SB_PDAC model with available mutational, transcriptome and proteomic data for human pancreatic cancer implicates these genes in disease progression. We use organoids, patient-derived cell lines and novel mouse models to delineate the biological context in which defined drivers cooperate to confer metastatic potential to pancreatic cancer cells and to investigate their participation in metastatic maintenance. The overall goal of our research is to identify biological vulnerabilities of pancreatic cancer metastases that may be harnessed for therapeutic targets.
We have also statistically identified a number of intriguing co-occurrence relationships between drivers in the SB_PDAC model and a high degree of intra-tumor heterogeneity (ITH). We are further investigating how ITH contributes to cancer progression. Using cells isolated from SB_PDAC tumors, we can enrich for sub clones that cooperate to drive primary tumors and resulting metastases in transplantation models. SB insertions tag tumor clones; using the SBCapSeq analysis platform we can identify the drivers present within the sub clones and explore the evolutionary relationships among primary and metastatic clones.