Overview
Tumors can be viewed as complex and dynamic ecosystems, where evolving populations of genetically and phenotypically heterogeneous cancer cells are engaged in dynamic reciprocal interactions with different types of non-malignant cells and non-cellular components of tumor microenvironment. Our lab seeks to understand the spatiotemporal dynamics of the tumor ecosystem, especially its response during the adaptation to anti-cancer therapies in space in time. Using this knowledge, we seek to devise therapies that aim beyond short term responses, by disrupting the ability of tumors to survive and adapt.
Integrating multiple sources of therapy resistance.
Despite the remarkable progress in identifying molecular mechanisms of therapy resistance, the progress in improving therapeutic outcomes has been relatively modest, with a partial exception of immunotherapy developments. The major obstacle to the therapeutic progress is the issue of intra-tumor heterogeneity of resistance mechanisms. If multiple resistant subpopulations are present in the same tumor, elimination of any single one can only achieve a temporal response. However, this is not the only issue. Less appreciated is the challenge posed by the complexity of resistance phenotypes. Even at the level of a single tumor cell, resistance might not be fully explainable by a single mutational or expression level change. Instead, it often integrates a contribution of multiple mutational and epigenetic mechanisms, as well as therapy-protective interactions with systemic and micro- environments. Therefore, even for a patient-specific case, adequate understanding of therapy resistance requires consideration of all relevant "building blocks" of resistance, as well as tumor heterogeneity. Our lab aims at gaining this knowledge by designing new methodological tools and striving to develop conceptual frameworks capable of incorporating multiple causes of resistance in order to provide adequate input for therapeutic decision making.

Disrupting the drivers of cancer evolution.
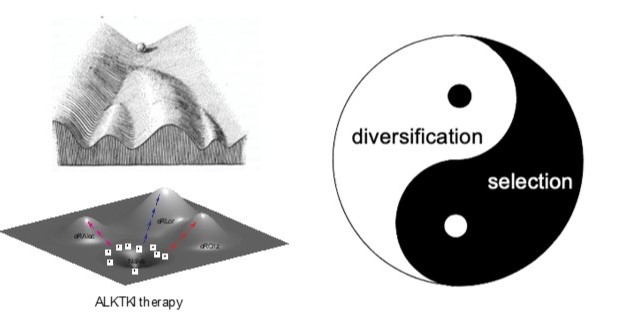
The evolution of resistant phenotypes can be conceptualized as trajectories traversing both epigenetic and adaptive landscapes. This process is driven by Darwinian interplay between diversification of (semi)heritable tumor cell phenotypes and fitness-based selection. Critically, diversification is a continuous process rather than a one time event. In addition to stochastic mutational processes, diversification of resistance-related traits is mediated by plasticity-mediated changes in gene expression (as long as these changes are at least partially heritable) and heterogeneity in tumor microenvironment. While cancer evolution is commonly assumed to be strictly clonal, our studies indicate that spontaneous cell fusions enable neoplastic populations to recombine mutations acquired by different clonal lineages, thus providing a parasexual mechanism. Suppression of diversification mechanisms and modulation of selection forces experienced by neoplastic populations might be needed to move from maximizing short term responses to achieving lasting improvements in long term therapeutic outcomes.
Development of evolutionary-informed therapeutic strategies.
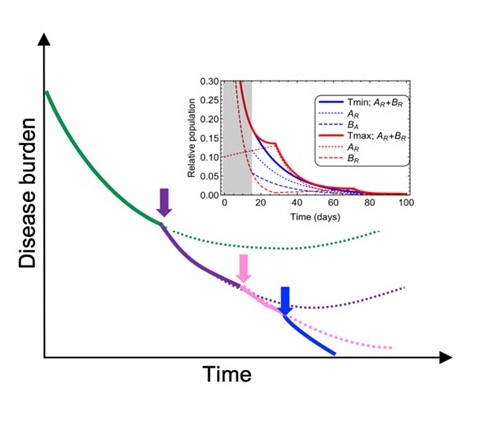
Given the challenges posed by the issues of intra-tumor heterogeneity and complexity of therapy-resistance phenotypes, focusing on a single molecular mechanism at a time is unlikely to solve the challenges of therapy persistence and resistance. Achieving more than incremental advances might require simultaneous consideration of tumor heterogeneity and the complexity of resistance phenotypes in space and time. Our lab is exploring several flexible strategies that aim to account for the complexity and the dynamic nature of therapy resistance by balancing preemptive and reactive adaptive therapy adjustments.